![]()
Dielectrics
We calculate MDMD.
[1]:
import time
import sys
import numpy as np
import matplotlib.pyplot as plt
from pathlib import Path
import glob
Some configurations if we are in a google colab environment:
[14]:
IN_COLAB = 'google.colab' in sys.modules
HAS_NEWANALYSIS = 'newanalysis' in sys.modules
if IN_COLAB:
if not'MDAnalysis' in sys.modules:
!pip install MDAnalysis
import os
if not os.path.exists("/content/mdy-newanalysis-package/"):
!git clone https://github.com/cbc-univie/mdy-newanalysis-package.git
!pwd
if not HAS_NEWANALYSIS:
%cd /content/mdy-newanalysis-package/newanalysis_source/
!pip install .
%cd /content/mdy-newanalysis-package/docs/notebooks/
Next we import MDAnalysis and functions needed from newanalysis.
[8]:
try:
import MDAnalysis
except ModuleNotFoundError as e:
if not IN_COLAB:
!conda install --yes --prefix {sys.prefix} MDAnalysis
import MDAnalysis
else:
print("Probably the cell above had a problem.")
raise e
try:
from newanalysis.correl import correlateParallel
from newanalysis.helpers import dipByResidue
from newanalysis.functions import atomsPerResidue, residueFirstAtom
except ModuleNotFoundError as e:
if not IN_COLAB:
print("Install newanalysis fist.")
print("Go to the newanalysis_source folder and run:")
print("conda activate newanalysis-dev")
print("pip install .")
else:
print("Probably the cell above had a problem.")
raise e
Preprocessing
Next we create our MDAnalysis Universe:
[9]:
base='../../test_cases/data/emim_dca_equilibrium/'
psf=base+'emim_dca.psf'
#Check PSF:
if np.array_equal(MDAnalysis.Universe(psf).atoms.masses, MDAnalysis.Universe(psf).atoms.masses.astype(bool)):
print("Used wrong PSF format (masses unreadable!)")
sys.exit()
u=MDAnalysis.Universe(psf,base+"emim_dca.dcd")
skip=20
dt=round(u.trajectory.dt,4)
n = int(u.trajectory.n_frames/skip)
if u.trajectory.n_frames%skip != 0:
n+=1
Now we do the selections
[10]:
sel_cat1 = u.select_atoms("resname EMIM")
ncat1 = sel_cat1.n_residues
mass_cat1 = sel_cat1.masses
charge_cat1 = sel_cat1.charges
com_cat1 = np.zeros((n,ncat1,3),dtype=np.float64)
mdcat1 = np.zeros((n,3),dtype=np.float64)
apr_cat1 = atomsPerResidue(sel_cat1)
rfa_cat1 = residueFirstAtom(sel_cat1)
print("Number EMIM = ",ncat1)
sel_an1 = u.select_atoms("resname DCA")
nan1 = sel_an1.n_residues
mass_an1 = sel_an1.masses
charge_an1 = sel_an1.charges
com_an1 = np.zeros((n,nan1, 3),dtype=np.float64)
mdan1 = np.zeros((n,3),dtype=np.float64)
apr_an1 = atomsPerResidue(sel_an1)
rfa_an1 = residueFirstAtom(sel_an1)
print("Number DCA = ",nan1)
Number EMIM = 1000
Number DCA = 1000
Analysis
[11]:
md = np.zeros((n,3),dtype=np.float64)
ctr=0
start=time.time()
print("")
for ts in u.trajectory[::skip]:
print("\033[1AFrame %d of %d" % (ts.frame,u.trajectory.n_frames), "\tElapsed time: %.2f hours" % ((time.time()-start)/3600))
# efficiently calculate center-of-mass coordinates
coor_cat1 = np.ascontiguousarray(sel_cat1.positions,dtype='double')
coor_an1 = np.ascontiguousarray(sel_an1.positions,dtype='double')
com_an1 = sel_an1.center_of_mass(compound='residues')
com_cat1 = sel_cat1.center_of_mass(compound='residues')
mdcat1[ctr] += np.sum(dipByResidue(coor_cat1,charge_cat1,mass_cat1,ncat1,apr_cat1,rfa_cat1,com_cat1),axis=0)
mdan1[ctr] += np.sum(dipByResidue(coor_an1,charge_an1,mass_an1,nan1,apr_an1,rfa_an1,com_an1),axis=0)
#Alternatively, use
#com_an1 = centerOfMassByResidue(sel_an1,coor=coor_an1,masses=mass_an1,apr=apr_an1,rfa=rfa_an1)
#com_cat1 = centerOfMassByResidue(sel_cat1,coor=coor_cat1,masses=mass_cat1,apr=apr_cat1,rfa=rfa_cat1)
#mdcat1[ctr] += np.sum(dipoleMomentByResidue(sel_cat1,coor=coor_cat1,charges=charge_cat1,masses=mass_cat1,com=com_cat1,apr=apr_cat1,rfa=rfa_cat1),axis=0)
#mdan1[ctr] += np.sum(dipoleMomentByResidue(sel_an1,coor=coor_an1,charges=charge_an1,masses=mass_an1,com=com_an1,apr=apr_an1,rfa=rfa_an1),axis=0)
md[ctr] += mdcat1[ctr]+mdan1[ctr]
ctr+=1
Frame 0 of 100 Elapsed time: 0.00 hours
Frame 20 of 100 Elapsed time: 0.00 hours
Frame 40 of 100 Elapsed time: 0.00 hours
Frame 60 of 100 Elapsed time: 0.00 hours
Frame 80 of 100 Elapsed time: 0.00 hours
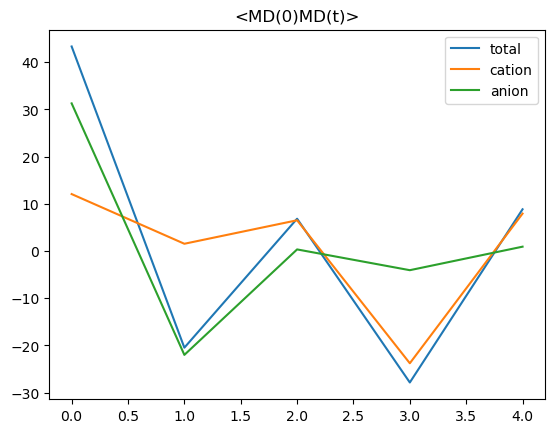
Calculate <MD(0)MD(t)>
[12]:
print("calculating <MD(0)MD(t)>")
# total
md = np.ascontiguousarray(md.T)
md0mdt = np.zeros(n)
correlateParallel(md,md,md0mdt,ltc=1)
#f1 = open("md0mdt.dat",'w')
#for i in range(len(md0mdt)):
# f1.write("%5.5f\t%5.5f\n" % (i*skip*dt, md0mdt[i]))
#f1.close()
# cations
mdcat1 = np.ascontiguousarray(mdcat1.T)
md0mdt_cat = np.zeros(n)
correlateParallel(mdcat1,md,md0mdt_cat,ltc=1)
#f1 = open("md0mdt_emim.dat",'w')
#for i in range(len(md0mdt)):
# f1.write("%5.5f\t%5.5f\n" % (i*skip*dt, md0mdt[i]))
#f1.close()
# anions
mdan1 = np.ascontiguousarray(mdan1.T)
md0mdt_an = np.zeros(n)
correlateParallel(mdan1,md,md0mdt_an,ltc=1)
#f1 = open("md0mdt_dca.dat",'w')
#for i in range(len(md0mdt)):
# f1.write("%5.5f\t%5.5f\n" % (i*skip*dt, md0mdt[i]))
#f1.close()
calculating <MD(0)MD(t)>
Plot the results
[13]:
time = np.arange(ctr*dt)
plt.title("<MD(0)MD(t)>")
plt.plot(time,md0mdt, label="total")
plt.plot(time,md0mdt_cat, label="cation")
plt.plot(time,md0mdt_an, label="anion")
plt.legend();