Theoretical background
\(\texttt{TRANSFORMATO}\) [1] is a python package designed to calculate either relative binding free energies (RBFE) or relative solvation free energies (RSFE) between two similar ligands. The two ligands are the two physical endstates which are mutated to a common core structure.
For that reason, \(\texttt{TRANSFORMATO}\) first searches for the common core (Common Core approach). All atoms which are not part of this CC and thus differ between the two ligands are then turned off step-wise. First, electrostatic contributions of the non-CC atoms are turned off, followed by turning off the Lennard-Jones (LJ) interactions of the non-CC atoms. For the non-hydrogen atoms this is done on an atom-by-atom basis (Serial-Atom-Insertion approach). This is done in both environments, for RSFE; ligand in vacuum and ligand solvated in water. For RBFE; ligand solvated in water and ligand solvated in water and bound to the receptor (protein).
\(\texttt{TRANSFORMATO}\) will create intermediate state folders to connect each ligand with its common core structure. Each folder will contain a plain MD simulation, which can be run individually from all other simulations. Finally, for each ligand the free energy according to the common core structure can be calculated. Since both common core structures are the same, the resulting free energy difference \(\Delta\Delta G^{bind}_{L1\rightarrow L2}\) can be obtained.
Interplay with CHARMM-GUI
\(\texttt{TRANSFORMATO}\) works closely together with CHARMM-GUI. For starting RBFE simulations, one needs output from the CHARMM-GUI solution builder.
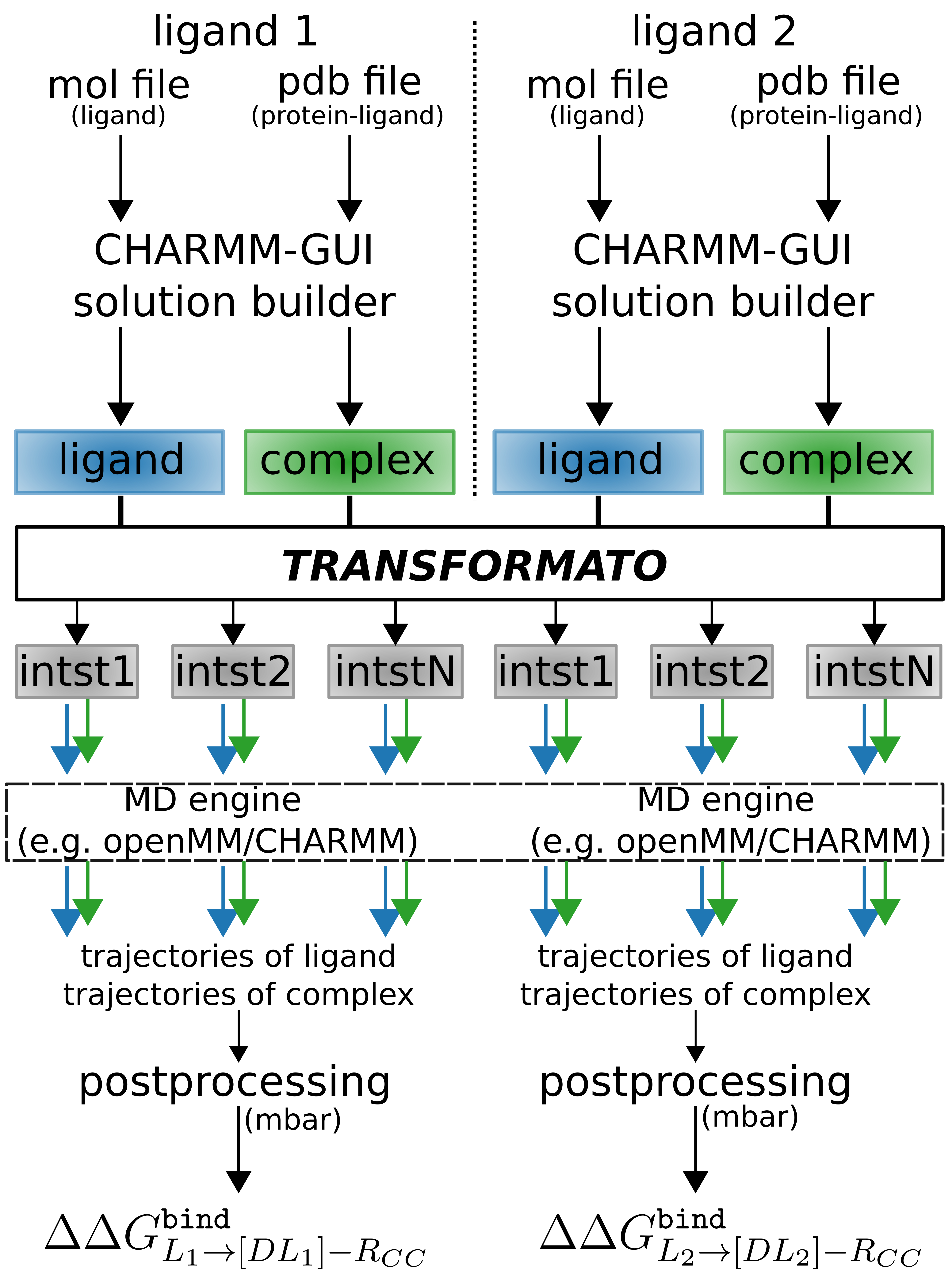
Figure: Interplay between \(\texttt{TRANSFORMATO}\) and CHARMM-GUI.
The common-core approach
With standard alchemical FES, the two endpoints are generally transformed directly into each other, with nonbonded forces (electrostatics) scaled according to the coupling parameter \(\lambda\), and bonded LJ interactions being turned off on an atom by atom basis. [3].
With \(\texttt{TRANSFORMATO}\) however, the structures are not transformed into each other directly. Instead, each structure is transformed into a ‘common core structure’, a common topology of both systems (ideally - and usually - the maximum common topology between systems), with the free energy being the sum of free energies necessary to reach the common core:
\(|DL_1|\) and \(|DL_2|\) refer to the non-CC (dummy region) of Endstate 1 and 2, respectively. \(R_{CC}\) refers to the common core.
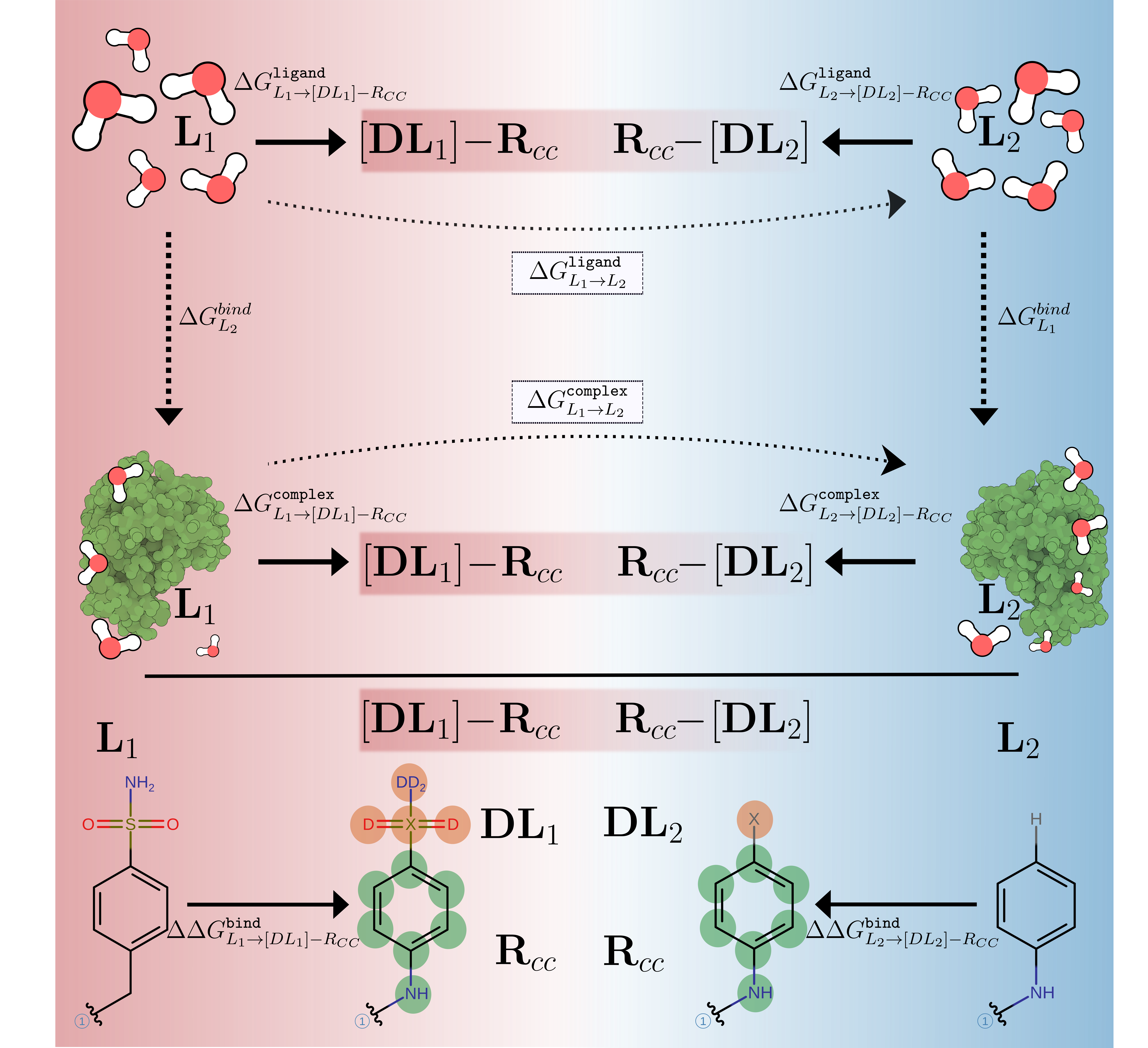
Figure: Alchemical pathway as implemented in the common core approach. Free energies are calculated relative to a common core structure in the two environments (for RBFE: ligand solvated in water and ligand bound to a protein and solvated in water). This common core structure of the two ligands differs only in the number of dummy atoms, thus, the contribution cancels out when calculating relative free energies.
Serial-Atom-Insertion approach
Lennard-Jones interactions are turned off on an atom-by-atom basis. This means that LJ parameters are either fully interacting (1) or non-interacting (0). For the heavy atoms (non hydrogen atoms) the interaction is turned off for each atom one by one.
Citations, License and Contact
Transformato is released under the MIT License. For further information, visit the repository license page.
Acknowledgements
Apart from the default python packages, \(\texttt{TRANSFORMATO}\) relies in large parts on the following packages:
MDAnalysis[5] [6] : Used for general purpose protein and ligand analysis, atom selection and especially trajectory analysis.
Numpy[7] : Well, it is numpy. We use it for math.
parmed[9] : Protein structure manipulations.
pymbar[8] : mBAR analysis.
RDKit: Visualisation of molecular structures.
References